
Alkane Dehydrogenation
By combining Density Functional Theory (DFT) calculations and microkinetic modeling (MKM), we unraveled the complex mechanisms involved in alkane dehydrogenation chemistries, revealing new insights at the atomic level. We developed a computational framework for quickly identifying effective catalysts through descriptor-based screening. In applying this framework, we discovered novel aluminum nitride catalysts with improved activity, promising more sustainable and cost-effective chemical processes. Our research represents a major leap forward in catalysis, with the potential to impact various industries.
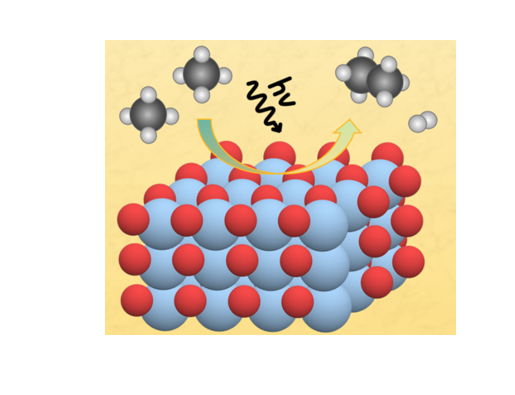
Nonoxidative Coupling of Methane (NOCM)
We explored the potential of titanium dioxide (TiO₂) as a catalyst for converting methane, a key component of natural gas, into useful chemicals. Through extensive kinetic Monte Carlo simulations, we assessed TiO₂'s performance and uncovered several important findings. Our research revealed that light and heat work together synergistically to enhance NOCM into ethane and hydrogen on TiO₂. This research, conducted in collaboration with Prof. Henrik Grönbeck from Chalmers University of Technology in Sweden, contributes valuable insights for developing more efficient and sustainable chemical processes.
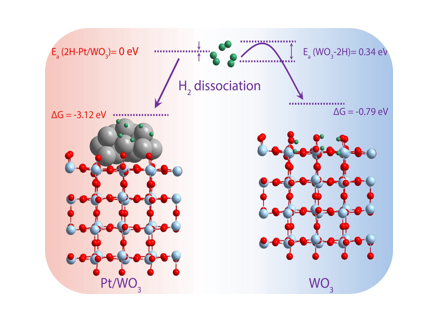
Hydrogen Spillover
We studied how hydrogen interacts with reducible metal oxides like tungsten oxide, using advanced multiscale modeling techniques. This first-principles-based approach allowed us to gain a detailed understanding of the mechanisms at play, paving the way for better manipulation and optimization of catalytic processes involving hydrogen. Conducted in collaboration with Dr. Ethan J. Crumlin from Lawrence Berkeley National Laboratory, this research offers critical insights that could revolutionize catalytic hydrogen-related transformations, leading to more efficient and sustainable industrial applications.
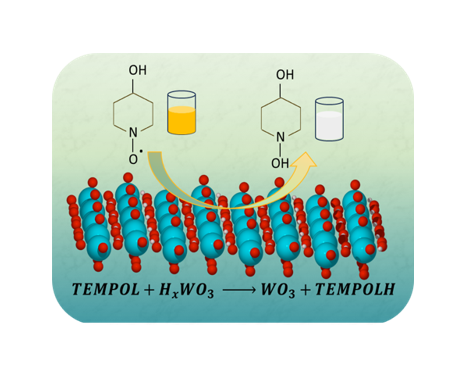
Electrochemical Looping for TEMPOL Hydrogenation
We examined the electrochemical looping hydrogenation of 4-hydroxy TEMPO on tungsten oxide bronzes. By delving into the mechanisms behind this reaction, we gained valuable insights into how these materials facilitate the conversion to 4-hydroxy TEMPO-H. This research, conducted in collaboration with Prof. James R. McKone from the University of Pittsburgh, advances our understanding of electrochemical looping processes, offering potential for more efficient hydrogenation techniques in the future.
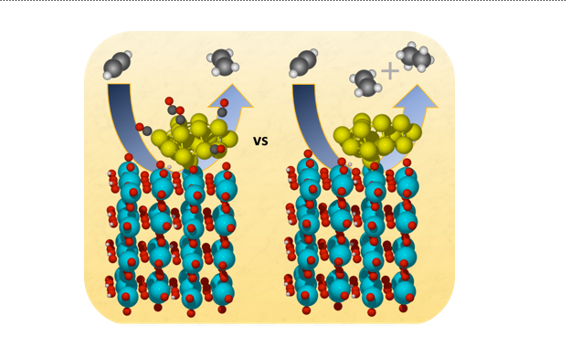
Acetylene Hydrogenation
Using DFT calculations, we studied the hydrogenation of acetylene to ethylene and ethane on Pt-decorated tungsten oxide with and without surface-bound carbon monoxide. Interestingly, the presence of CO increased the rate of ethylene production and reduced over-hydrogenation to ethane. This research, conducted in collaboration with Prof. Götz Veser from the University of Pittsburgh, provides valuable insights into optimizing hydrogenation reactions, highlighting the potential for using less pure hydrogen streams in industrial processes.
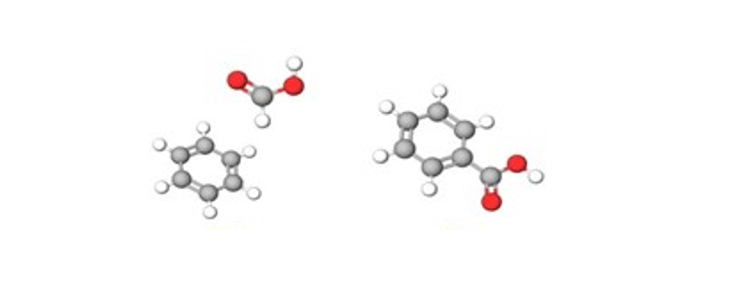
Direct Carboxylation of Arenes
We investigated the direct carboxylation of benzene to produce benzoic acid on copper-nickel nanoparticles, using formic acid as a carboxylating agent. Through DFT calculations, we revealed that the carboxylation process involves activating the C-H bonds in both benzene and formic acid. This study, conducted in collaboration with Prof. Shaikh M. Mobin from the Indian Institute of Technology-Indore, offers new insights into sustainable chemical production.
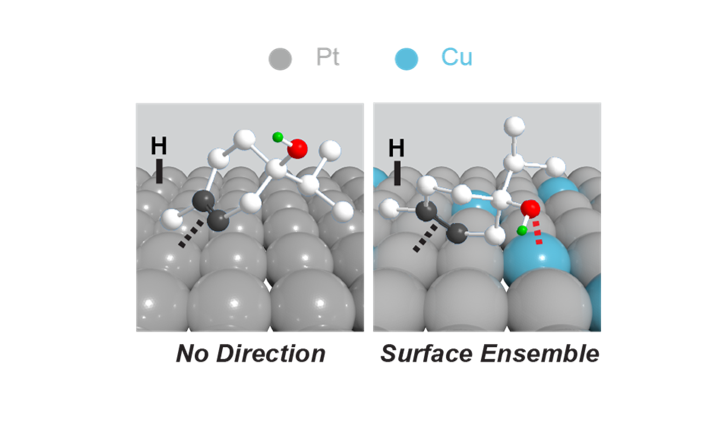
Directed Hydrogenation of Terpinenol
Using DFT calculations, we assessed the high conversion and selectivity observed in the hydrogenation of terpinen-4-ol on multimetallic surfaces, showcasing how the hydroxyl groups direct the reaction to achieve such results. This study, conducted in collaboration with Prof. Christina Li from Purdue University, provides deeper insights into optimizing directed hydrogenation reactions, which provide access to highly functionalized and diastereomerically pure products.
Create Your Own Website With Webador